


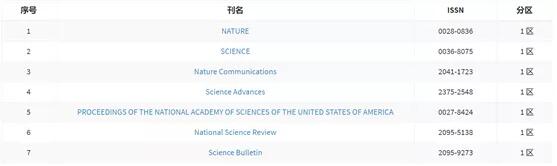
会议
- 会议
- 资讯
乔世璋教授Angew Chem: 谱学+计算——必(Bi)成其美(Mg)
更新时间:2020-08-19
【研究亮点】
1. 提出了Mg/Bi电池体系新储镁机理,通过一系列原位、非原位同步辐射光谱(近边X-射线吸收精细结构、X-射线粉末衍射)和DFT理论计算证实;
2. 揭示镁铋合金化中间体(MgBi)优异物化性能。DFT理论计算显示,MgBi中间体存在低的体积膨胀率、优良动力学性能和高导电性,有助于电极材料电化学性能提高。
3. 柔性、自支撑、耐腐蚀多孔Bi负极材料设计。介孔结构设计,可缓解合金化反应大的体积膨胀和提高离子扩散动力学性能;柔性碳上的活性材料Bi作为电极,体系简单,适合储镁机理研究;碳集流体,耐镁电解液腐蚀。精细的电极结构设计显著提高了材料电化学性能。在应用有机非混合离子镁电解液的Mg/Bi体系中,表现出优异的电化学性能。
【研究背景】
镁是地壳中第八大丰富元素。镁的广泛可用性和可及性、高体积能量密度(3833mAhcm-3)、无枝晶安全、多价离子等特性,使其成为继锂离子电池后研究的新热点。但是,镁金属负极和传统碳酸酯类电解液的不兼容性,很大程度限制其实际发展和应用。当前,可使用的有机镁电电解液以醚类电解液为主,该类电解液具有强腐蚀性,空气、水敏感性和较差的阳极稳定性。研究开发可在传统电解液中稳定、可逆工作、低电压的金属电极(Bi)替代Mg,将为高电压、高能量密度镁电池的开发提供新方向。
【研究思路剖析】
当前,镁电的替代负极主要是合金类材料,包括Bi、Sn、Sb、Pb及对应多元合金等。合金类电极主要的问题存在于合金化体积膨胀大,导致活性材料粉碎、脱落,失去电活性。总结前期报导,其中Bi电化学性能相对稳定,体积容量高,受到研究人员关注。但是,Bi相对大的体积膨胀(我们理论计算得到为204%,Mg3Bi2)以及二价Mg2+在Bi宿体缓慢的固相扩散,很难实现电池长循环和高的倍率性能。从材料合成角度制备介孔纳米Bi电极,可减小体积膨胀应力和缩短离子扩散路径,最终提高材料结构稳定性和改善动力学性能。对Bi电极反应机理探索,在Li/Bi、K/Bi、Na/Bi电池体系中,合金化皆是多步反应过程。已有的Mg/Bi报道中,Mg合金化过程只是一步反应生成Mg3Bi2。为进一步研究验证其反应机理,作者利用高精度、高分辨率的同步辐射测试手段,对反应机理深入解析,结合理论计算研究,提出Mg/Bi两步反应机理,探索出影响Bi合金电极材料性能的一些关键因素。
【图文解析】

图1. 储镁机理分析。a)非原位Raman分析;b)同步辐射NEXAFS谱图;c)同步辐射原位XRD;d)Mg/Bi 合金化过程体积变化理论计算
要点1:近边X-射线吸收精细结构谱 (NEXAFS)提供可靠的电子和中心原子化学态信息。作者通过同步辐射非原位NEXAFS谱分析,放电初始到末态,Mg K边位置变化出现中间过渡态;进一步分析,利用同步辐射原位XRD探测,发现MgBi中间相。提出Mg/Bi合金反应过程是一个两步反应:
Bi + Mg2+ + 2e‒↔ MgB (1)
2MgBi + Mg2+ + 2e‒↔ Mg3Bi2 (2)
理论计算Mg/Bi合金反应体积变化,中间相MgBi具有较小的体积变化率,反应生成最终相体积变化为204%。由于Bi较大的体积变化,因此对Bi材料的结构设计提出了更高的需求。
_副本.jpg "640 (1)_副本.jpg")
图2. 电极材料形貌结构和晶体结构变化
要点2:柔性、自支撑介孔Bi纳米片(p-Bi NS)平均孔尺寸17.6 nm,电解液可快速有效浸润材料。非原位TEM分析,放电镁化过程,多孔结构可有效容纳体积膨胀,高分辨TEM可捕捉到MgBi 晶相,证实中间相的存在。充电脱镁过程,材料结构保持完好。这证明了该结构设计的优异性,可有效缓解体积膨胀,保持结构稳定性。
图3. Mg/Bi电池体系电化学性能。a)循环伏安测试;b)充放电压曲线;c)倍率性能测试;d)循环稳定性测试;e)电化学阻抗谱拟合结果
要点3:p-Bi NS和商业Bi电极电化学性能对比。通过精细结构设计的p-Bi NS电极,能发挥出更高的电池容量、更好的循环性能和倍率性能。放电曲线和阻抗分析,p-Bi NS表现出极小的形核过电位,低的界面电阻。这样的电极材料设计,可提高材料活性,有助于提高电化学性能,同时利于电极反应机理的研究。
图4.分子动力学模拟。a)Mg扩散路径;b)不同扩散路径能垒图;c)AIMD模拟
要点4:分子动力学模拟,从原子角度说明Mg2+在Bi基体中的迁移过程,根据理论扩散能垒分析,Mg2+主要沿Bi (110) 晶面迁移;Mg可在较短时间与Bi相互作用,最终形成MgxBi合金。
_副本.jpg "640 (4)_副本.jpg")
图5. DFT理论计算。a)放电曲线理论模拟;b)相变反应能垒;c)不同合金态电子态密度分析;d)示意图。
要点5:理论计算放电平台0.25V和0.21V,分别对应生成MgBi和Mg3Bi2。两平台数值相差很小,因此实际放电曲线很难区分,近似一个平台的”假象”。相变反应能垒计算,经历中间相的两步反应,能垒更低。合金化过程,更倾向于通过两步反应。MgBi总电子态密度在费米能级处有分布且能量带隙为0,具有金属特性; Mg3Bi2为半导体。由此,Mg/Bi体系发生两步反应,中间相(MgBi)的生成,对电极材料电化学性能有很大影响。
【全文小结】
针对Mg/Bi电池体系,作者提出了新的两步反应机理。反应中间相(MgBi)为金属导体,可提高材料导电性;小的体积膨胀率使其可作为体积变化缓冲物质;低的反应能垒,表现出良好的动力学性能。通过电极材料结构设计,可提高材料结构稳定性和反应动力学性能。作者制备出柔性、自支撑耐腐蚀介孔Bi纳米片,这样的材料设计,在应用有机非混合离子镁电解液的Mg/Bi体系中,表现出最优异的电化学稳定性。
Xin Xu, Dongliang Chao, Biao Chen, Pei Liang, Huan Li, Fangxi Xie, Kenneth Davey, and Shi-Zhang Qiao, Revealing the Magnesium Storage Mechanism in Mesoporous Bismuth via Spectroscopy and Ab Initio Simulation, Angew. Chem. Int. Ed. 2020, DOI:10.1002/anie.202009528
来源:能源学人


















-
.png)
 顾问老师
顾问老师扫码添加顾问老师

-
.png)
 微信客服
微信客服扫码添加平台客服

-
.png)
 客服电话
客服电话电话:13922151049
-
.png)
 公众号
公众号关注AEIC公众号查看更多资讯

